PROVE SEMI-DISTRUTTIVE e DISTRUTTIVE per la DIAGNOSI delle COSTRUZIONI
IL METODO DEI MARTINETTI PIATTI
Il metodo (1) è largamente impiegato nelle strutture murarie per la determinazione di due grandezze:
a) tensione attuale in una zona della muratura
b) modulo elastico statico.In alcuni casi si può anche determinare il carico di rottura per compressione di un determinato concio murario.
La prova, a rigore, non è proprio non-distruttiva soprattutto quando si giunge alla rottura per compressione del concio in sito: tuttavia, rispetto ad un’analoga prova che potrebbe essere eseguita in laboratorio prelevando una porzione muraria mediante carotaggio o asportazione di un concio, essa comporta indubbiamente un danno irrilevante alla struttura originale, soprattutto se si può effettuare la riparazione in sito. Inoltre, essa presenta il vantaggio di indagare una porzione significativa di materiale, e di valutare il comportamento medio della struttura indipendentemente dal fatto che essa sia omogenea o, come spesso avviene, eterogenea (malta, mattoni e/o pietra).
Per la determinazione della tensione attuale si prendono in considerazione due punti (A e B) la cui distanza è preliminarmente misurata con un estensimetro rimovibile (Fig.1).

Dopo aver effettuato un taglio (di circa 1 cm), si inserisce un martinetto piatto opportunamente sagomato (cilindrico, rettangolare, ecc.) in funzione della geometria della muratura. Dopo aver effettuato il taglio nella muratura, e prima di inserire il martinetto, la distanza tra i punti A e B è variata (per aver rimosso con il taglio la tensione esistente) ed è diventata quella relativa tra A’ e B’. Si applica una pressione attraverso il martinetto fino a ripristinare la distanza tra i punti A e B che esisteva in corrispondenza della tensione originale: dalla pressione misurata e dalla geometria del martinetto si calcola la tensione che agisce normalmente alla sezione del taglio.
Per la misura del modulo elastico statico, o più esattamente della curva sforzo-deformazione per il concio di muratura da indagare, si effettuano due tagli paralleli nei quali vengono alloggiati due martinetti piatti (Fig. 2).

I punti A, A’ e B, B’ individuano una coppia di segmenti la cui distanza varia allorquando i martinetti piatti vengono caricati con olio in pressione. Dal valore della pressione (P) applicata si calcola la sollecitazione di compressione (σc), mentre la variazione Δl misurata tra A e A’, o tra B e B’ consente di calcolare la deformazione unitaria ε = Δl/l0, in corrispondenza di ogni σc lungo la direzione di applicazione del carico e quella ortogonale.
Riportando i valori di σc in funzione di ε calcolato per le distanze AA’ e BB’ (Fig. 3) si ricava la curva di sforzo-deformazione: la pendenza della curva σc-ε(AA’) consente di calcolare il modulo elastico statico, mentre il rapporto εBB’/εAA’ consente di determinare il modulo di Poisson.
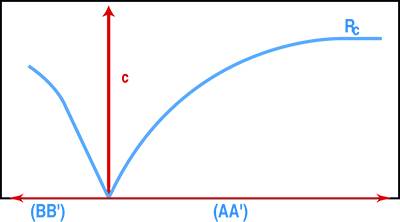
Se la pressione dell’olio che alimenta il martinetto aumenta fino alla rottura del concio, si arriva a determinare direttamente la resistenza meccanica (Rc), la quale però è molto spesso valutata per estrapolazione della curva σc-ε senza arrivare completamente alla rottura del concio (Fig.4) per consentire una più semplice riparazione in sito della muratura dopo aver terminato la misura.
PROVE DISTRUTTIVE
Le prove distruttive consistono fondamentalmente in prove di laboratorio di carattere prevalentemente chimico, mineralogico, e fisico; talvolta esse includono anche prove di carattere meccanico molto simili alle prove non-distruttive (modulo elastico dinamico mediante ultrasuoni, curva sforzo-deformazione fino alla rottura del provino).
Tabella 1 - Principali prove distruttive
• Analisi chimica
• Analisi per diffrazione dei raggi X
• Analisi termica
• Spettrofotometria a raggi IR
• Microscopia elettronica
• Assorbimento di liquidi
• Porosimetria a mercurio
• Permeabilità all’acqua
• Adsorbimento di gas
• Prove meccaniche
Escludendo quelle di carattere meccanico già discusse nella sezione delle prove non distruttive, le prove distruttive che verranno esaminate sono riportate nella Tabella 1. Val la pena di sottolineare che molto spesso si identificano le prove distruttive di laboratorio con l’analisi chimica, la quale invece, di per sé e da sola, non è in grado di fornire elementi significativi per l’emissione di una diagnosi se non è accompagnata dalle altre prove non distruttive di carattere mineralogico o fisico.
ANALISI CHIMICA ELEMENTALE
L’analisi chimica tradizionale di un solido (come un mattone, una malta, una pietra o un frammento di calcestruzzo) consiste nel disciogliere con adeguati solventi (acidi, basi, ecc.) il campione in acqua e nell’analizzare gli ioni presenti nella soluzione. Questo tipo di analisi è definita analisi chimica elementale, poiché consente di determinare la composizione del materiale in termini di percentuale degli elementi (Ca, Na, K, ecc.).
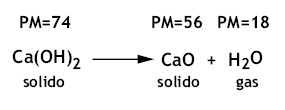
Per esempio, nel caso di un solido costituito d un unico sale come il CaSO4, la dissoluzione avviene nel modo seguente:

Il chimico analista è in grado di stabilire quanto calcio e quanto solfato sono presenti come ioni nella soluzione acquosa e quindi può facilmente calcolare le percentuali di calcio e di solfato presenti nel solido esaminato conoscendo la quantità di solido e di solvente impiegati.
Se il solido è costituito da una miscela di due sali, come il CaSO4 ed il K2CO3, l’attacco dissolvente porterà in soluzione i seguenti ioni: Ca+2, SO4-2, K+, CO3-2.
In questo caso il chimico analista sarà in grado di determinare la quantità dei singoli ioni nella soluzione acquosa, ma non è più in grado di stabilire se nel solido di partenza erano presenti la miscela (I) oppure quella (II):

o addirittura miscele ancor più complesse come:

Tutte e sette le miscele di sali da (I) a (VII) potrebbero, una volta disciolte in acqua, in teoria corrispondente all’analisi chimica che ha portato alla determinazione dei quattro ioni: Ca+2, K+, SO4-2, CO3-2.
Stante questa difficoltà interpretativa, il chimico analista si limita a comunicare le percentuali dei quattro ioni. Per esempio, nel caso di una miscela costituita da 30% di CaSO4 e 70% di K2CO3, il risultato sarà:
Tutte e sette le miscele di sali da (I) a (VII) potrebbero, una volta disciolte in acqua, in teoria corrispondente all’analisi chimica che ha portato alla determinazione dei quattro ioni: Ca+2, K+, SO4-2, CO3-2.
Stante questa difficoltà interpretativa, il chimico analista si limita a comunicare le percentuali dei quattro ioni. Per esempio, nel caso di una miscela costituita da 30% di CaSO4 e 70% di K2CO3, il risultato sarà:
- Ca+2 = 8,8%
- SO4-2 = 21,2%
- K+ = 39,6%
- CO3-2 = 30,4%
Più frequentemente il risultato viene espresso sotto forma di percentuali di ossidi corrispondenti agli elementi (CaO, K2O, SO3, CO2) presenti e pertanto la composizione diventa:
- CaO = 12,4%
- SO3 = 17,6%
- K2O = 47,7%
- CO2 = 22,3%
DIFFRAZIONE DEI RAGGI X
L’analisi per diffrazione dei raggi X – detta anche XRD (X Ray Diffraction) – di un materiale solido viene effettuata sul campione stesso, in forma di polvere finemente macinata, senza doverlo sciogliere in acqua. Essa consente di avere informazioni addizionali che permettono di interpretare meglio il risultato dell’analisi chimica.
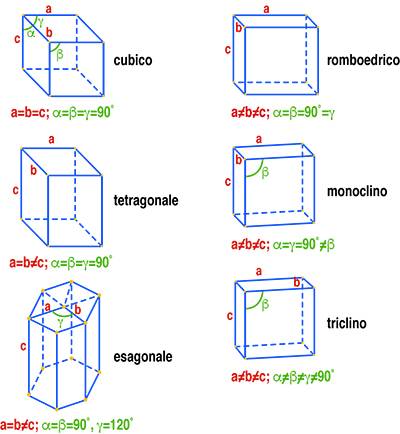
L’analisi per diffrazione dei raggi X è applicabile solo ai solidi cristallini nei quali gli atomi che compongono il materiale sono disposti in modo geometricamente ordinato. Nei solidi cristallini gli atomi occupano delle posizioni corrispondenti ai vertici di figure geometriche ben precise, quali quelle mostrate in Fig. 4 e denominate celle elementari.

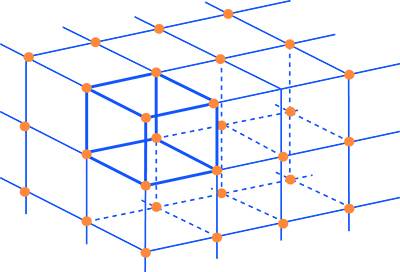
Traslando nelle tre direzioni dello spazio una cella elementare si può ottenere la rappresentazione cristallografica del solido. La Fig. 5 mostra il reticolo spaziale di un cristallo di forma tetragonale dove viene evidenziata la cella unitaria del prisma tetragonale.
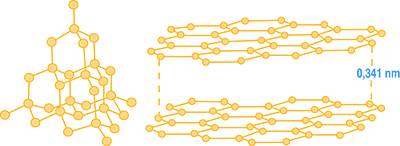
Gli atomi sono indicati dai punti corrispondenti ai vertici delle varie celle unitarie che compongono il reticolo, ma nella realtà sono come delle sfere quasi in contatto lasciando poco spazio tra loro. Nella Fig. 6 sono mostrate le rappresentazioni del diamante e della grafite, che sono due forme allotropiche: cioè si differenziano per la struttura cristallina dello stesso elemento (il carbonio) e che pertanto non sarebbero distinguibili attraverso la sola analisi chimica elementale.
Esistono solidi cristallini costituiti da atomi (per esempio il diamante o la grafite) o da ioni (per esempio il cloruro di sodio costituito da Na+ e Cl-). Per i prodotti inorganici, che costituiscono i materiali da costruzione, il reticolo cristallino è formato generalmente da ioni, mentre nei materiali metallici sono gli atomi a costituire il reticolo cristallino.
Non sempre gli atomi o gli ioni si dispongono in maniera ordinata ai vertici del reticolo cristallino. Talvolta, essi sono “disturbati” dal raggiungere questa situazione ordinata (per esempio da un raffreddamento troppo rapido del liquido in via di solidificazione) ed assumono allora una disposizione più o meno irregolare; si dice allora che il solido è amorfo.
Tra i prodotti amorfi più frequenti nel campo dei materiali solidi vi è la silice (SiO2). Essa può cristallizzare in diverse forme quali il quarzo α, il quarzo β, la cristobalite, la tridimite, ma può anche rimanere amorfa come avviene per esempio nelle pozzolane, o in altri minerali silicei come l’opale ed il calcedonio.
La Fig. 7 mostra i piani reticolari di uno stesso composto (per esempio SiO2) che è riuscito a cristallizzare (A) in confronto allo stesso composto che si presenta invece amorfo (B).

E’ evidente che nel caso dei prodotti amorfi diventa difficile individuare una serie di distanze reticolari caratterizzanti quel composto. La diffrazione dei raggi X, come si vedrà nel paragrafo successivo, consente di riconoscere solo un prodotto cristallino emettendo dei segnali in forma di picchi mentre presenta solo una banda diffusa per i materiali amorfi.
L’equazione di Bragg
Ogni piano reticolare si comporta rispetto ai raggi X esattamente come una riga in un reticolo di diffrazione per la luce visibile. Le posizioni delle righe spettrali, quando risultano dalla diffrazione della luce visibile di un reticolo a righe, dipendono dalla distanza fra le righe successive; analogamente, la diffrazione dei raggi X è determinata dall’intervallo fra i piani successivi. Si supponga che le rette AA, BB, CC, ecc. nella Fig.8 rappresentino un certo numero di piani reticolari in un reticolo spaziale, i cui punti siano gli atomi che costituiscono il cristallo.

Se un fascio parallelo di raggi X incide su un cristallo in modo che l’angolo d’incidenza sia θ, una parte del fascio, per esempio LM, viene riflesso in M lungo MN dagli atomi nel piano AA. D’altra parte, altri raggi, per esempio PQ, penetrano nel cristallo e vengono riflessi, per esempio lungo QN, dagli atomi nel piano BB; analogamente si hanno riflessioni sugli altri piani CC, ecc. Se la differenza tra il percorso LMN e quello PQN è eguale a un numero intero di lunghezze d’onda, i due fasci, da L e da P, si addizionano rinforzandosi dopo la riflessione e ne risulta un intenso fascio N diffratto.
Per individuare la situazione di sovrapposizione e di rinforzo dei raggi diffratti si tracci da M la perpendicolare MR ai piani di riflessione e la MS perpendicolare a QR. La differenza fra i percorsi LMN e PQN è eguale a QM – QS, e poiché QM = QR la differenza tra i percorsi è uguale ad SR. Dato che l’angolo SMR è uguale all’angolo d’incidenza θ, si deduce che SR è uguale a 2d sen θ, in cui d è la distanza fra due piani reticolari successivi. Perché si abbia un massimo di riflessione questa deve essere un numero intero (n) di lunghezze d’onda (λ); perciò la condizione, che si applica anche ai raggi riflessi dagli altri piani, è data dalla [1] nota come equazione di Bragg:
nλ = 2d sen θ [1]
Per un certo solido cristallino caratterizzato da una data serie di piani reticolari, e per un fascio di raggi X aventi una certa lunghezza d’onda nota, d e λ sono determinati; quindi l’intensità della diffrazione dipende dall’angolo d’incidenza θ. Aumentando gradualmente θ, (facendo variare l’angolo di incidenza dei raggi X rispetto al piano degli atomi) si trova una serie di posizioni, corrispondenti ad n = 1, 2, 3, 4, ecc., per le quali si verificano massimi di riflessione, separati da regioni in cui i vari raggi diffratti non sono in fase e quindi si annullano l’un l’altro in gran parte. Per mezzo di un cristallo è possibile, quindi, ottenere uno “spettro” di raggi X; i massimi di diffrazione vengono chiamati di primo, secondo, terzo, ecc. ordine secondo il valore di n. Da tale spettro, per mezzo dell’equazione di Bragg, è possibile determinare la distanza d che separa i piani reticolari. Questa applicazione (nota come cristallografia) ha consentito di caratterizzare i reticoli cristallini di molti minerali attraverso la determinazione delle varie distanze reticolari d. Nella pratica applicata alla diagnostica, invece, poiché sono ormai note le distanze reticolari dei vari prodotti cristallini, è possibile riconoscere questi prodotti attraverso lo spettro ai raggi X quando sono presenti in una miscela incognita.
Dal punto di vista analitico, cioè per arrivare alla individuazione di solidi cristallini in un materiale, viene impiegato il cosiddetto metodo delle polveri che consiste nel sottoporre a diffrazione dei raggi X il materiale da analizzare ridotto in polvere finissima per macinazione (Fig.9). Nella polvere i cristalli sono orientati casualmente in tutte le direzioni e pertanto un certo numero di essi avranno sempre i loro piani reticolari nella direzione esatta affinché sia applicabile l’equazione di Bragg.

Per esempio, la Fig. 10 mostra il diffrattogramma dei raggi X ottenuto per il quarzo, mentre nella Fig. 11 è riportato il diffrattogramma della silice amorfa chimicamente identica (SiO2) al quarzo. In entrambi i diffrattogrammi viene riportata l’intensità dei raggi diffratti (in colpi al secondo: c/s) in funzione dell’angolo 2θ, misurato con un goniometro girevole.


Si può vedere che solo il quarzo, per la sua cristallinità, presenta dei picchi (corrispondenti ai massimi di riflessione dei raggi X) in corrispondenza di determinati angoli che sono caratteristici proprio del prodotto cristallino esaminato.
La silice amorfa, pur essendo chimicamente identica al quarzo, non presenta invece alcun picco proprio per l’assenza di piani reticolari distanziati con regolarità tra loro. Questo è uno dei limiti della diffrazione dei raggi X: il non potere riconoscere con questa tecnica, per l’assenza di picchi sul diffrattogramma, i prodotti solidi amorfi. Ciò significa che la diffrattometria dei raggi X non consente di distinguere tra un materiale costituito da quarzo solo, da un altro contenente quarzo e silice amorfa: in entrambi i casi verranno evidenziati solo i picchi del quarzo.
D’altra parte, l’altezza dei picchi, pur essendo proporzionale alla percentuale di prodotto cristallino, dipende anche da un così alto numero di parametri (grossezza dei grani cristallini, compattezza della polvere inserita nel portacampione, ecc.) che diventa molto difficile, se non con tecniche molto laboriose e sofisticate, arrivare ad una determinazione quantitativa precisa. Sebbene l’altezza dei picchi consenta solo una valutazione semi-quantitativa della composizione, la diffrazione dei raggi X resta uno dei metodi più efficaci per una valutazione qualitativa dei prodotti solidi cristallini presenti in un materiale.
Per esempio, essa consente facilmente di distinguere i sali cristallini presenti in una miscela, dalla cui analisi chimica non si sarebbe potuto arrivare alla identificazione dei singoli prodotti.
Nella Fig. 12 è mostrato il diffrattogramma di una miscela incognita di composti.

Questo diffrattogramma viene confrontato con quelli di vari composti puri già noti in letteratura. La comparazione consente di dedurre che la miscela dei composti è formata da CaCO3 ed NaCl. Questo confronto, che in realtà viene eseguito attraverso un programma computerizzato, consiste nel comparare i picchi presenti nel diffrattogramma della miscela di composizione incognita con quelli di tutti i composti noti fino ad attribuire qualsiasi picco (anche il più piccolo) ad uno presente in qualche composto noto.
Nella Fig. 13 la comparazione è limitata per chiarezza solo ai picchi più importanti che appartengono al CaCO3 ed all’NaCl.

In sostanza, il diffrattogramma di ciascun prodotto si presenta come una sorta di impronta digitale e il diffrattogramma della miscela incognita dei prodotti, anch’essa una impronta digitale ottenuta dalla sommatoria delle impronte di tutti i prodotti in essa contenuti, viene comparata con quella di tutti i composti noti (e dei quali si possiede lo spettro XRD) fino alla loro completa identificazione.
L’analisi per diffrazione dei raggi X è il mezzo più efficace per riconoscere la presenza di composti (purché cristallini e non amorfi) in un campione solido da identificare. Tuttavia, il mezzo non è molto adatto per risalire alle percentuali dei singoli composti se non in modo approssimativo. Da questo punto di vista l’analisi termica discussa nel prossimo paragrafo è più semplice come metodo per l’analisi quantitativa.
ANALISI TERMICA
segue ...